PENDAHULUAN
Menurut Setianingsih
(2008), talasemia merupakan penyakit genetik yang menyebabkan gangguan
sintesis rantai globin, komponen utama molekul hemoglobin (Hb). Talasemia
diturunkan secara autosomal resesif. Gejala yang paling ringan (bentuk
heterozigot) yang disebut dengan talassemia minor dan talassemia trait,
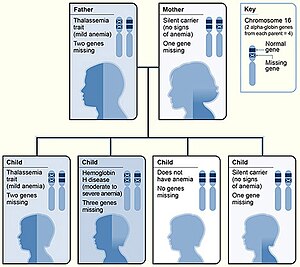
hingga yang berat yaitu talasemia mayor. jika kedua orang tua pembawa gen
talasemia, kemungkinan anak menderita talasemia sebesar 25%, pembawa gen
50% dan normal 25%.
Pola Genetik pada Talasemia-β

Talasemia-α
Talasemia mayor (sifat gen dominan) ditandai dengan kurangnya
kadar hemoglobin
Pasien akan tampak normal
saat lahir, namun di usia 3-18 bulan akan mulai terlihat adanya gejala anemia. Selain itu,
juga bisa mincul gejala lain seperti takikardi dan facies cooley yang merupakan ciri khas talasemia mayor dengan
tampilan batang hidung masuk ke dalam dan tulang pipi menonjol, akibat sumsum
tulang yang bekerja terlalu keras untuk mengatasi kekurangan hemoglobin.
Pada umumnya, penderiyta
talassemia mayor hars menjalani transfusi darah dan pengobatan seumur hidup. Seberapa
sering transfusi darah ini harus dilakukan tergantung berat ringannya penyakit.
Semakin berat penyakitnya, kian sering pula si penderita harus menjalani
transfusi darah.
Sedangkan pada talassemia
minor, individu hanya membawa gen penyakit ini, namun individu hidup normal,
tanda-tanda penyakit talassemia tidak muncul. Namun bila nantinya menikah
dengan individu talassemia minor, kemungkinan 25% anaknya menderita
talassemia mayor.
EPIDEMIOLOGI
Pada bagian anak Rumah
Sakit Cipto Mangunkusumo, prevalensi anemia pada kelompok orang dewasa tercatat
29% (defisiensi nutrisi), 31% (thalassemia), 10% (aplastik), 4%(hemolitik) dan
26% (penyebab lainnya). Tahun 1955 Li-Injo pertama kali melaporkan, HbE
merupakan kelainan yang paling sering ditemukan di berbagai etnik di Indonesia,
2.5%-13.2%. Di studi berikutnya prevalensi yang dilaporkan cukup bervariasi,
9.5%(bayi baru lahir), 22% (wanita hamil) dan 15.9-60% (pada atlet). Karier juga
bervariasi antara 6-10%.
Di RSCM sampai dengan
akhir tahun 2003 terdapat 1060 pasien talassemia mayor yang terdiri dari 52.5%
pasien thalassemia β homozigot, 46.2% thalassemia HbE, serta talassemia α
1.3%. sekitar 70-80 pasien baru datang tiap tahunnya. Pembawa sifat talassemia
β di Indonesia ditemukan lebih tinggi yaitu 3%-10%, pembawa sifat talassemia α
2.6%-11% dan pembawa hemoglobin E 1.5%-33%.
PATOFISIOLOGI
Ada beberapa jenis
hemoglobin yang disesuaikan dengan kebutuhan oksigen selama masa pertumbuhan,
mulai embrio, fetus sampai dewasa. Hemoglobin memiliki bentuk tetrametrik yang
sama, terdiri dari dua pasang rantai globin yang terkaint dengan heme. Heme terdiri
dari zat besi (Fe) sedangkan globin suatu protein yang terdiri dari rantai
polipeptida. Sintesa globin dimulai pada awal kehidupan masa embrio di dalam
kandungan sampai 8 minggu usia kehamilandan hingga akhir kehamilan. Organ yang bertanggung
jawab pada periode ini adalah hati, limpa dan sumsum tulang.
Pada thalassemia-β,
kelebihan rantai alfa mengendap pada membran sel eritrosit dan merupakan
prekursor yang menyebabkan penghancuran eritrosit yang hebat. Eritrosit yang
mencapai darah tepi memiliki inclusion bodies yang menyebabkan penghancuran di
limpa dan oksidasi membran sel akibat pelepasan heme dan denaturasi hemoglobin
dan penumpukan besi pada eritrosit.
Anemia pada thalassemia-β
terjadi akibat hancurnya eritrosit dan umur eritrosit yang pendek. Penimbunan eritrosit
yang hancur di limpa mengakibatkan terjadinya pembesaran limpa yang diikuti dengan
terperangkapnya leukosit dan trombosit sehingga menimbulkan gambaran
hipersplenisme.
DIAGNOSIS DAN PEMERIKSAAN
Terdapat empat diagnosis
utama pada penderita thalassemia, yatu :
1. Terdapat gambaran sel darah merah mikrositik yang banyak sehingga
nilainya jatuh kepada diagnosis anemia,
2.
Dari anamnesis terdapat riwayat keluarga yang menderita penyakit
yang sama,
3. Gambaran sel darah merah abnormal yakni mikrositik, acanthocytes
dan terdapat sel target, dan
4.
Untuk thalassemia-β, terdapat peningkatan hemoglobin α2 atau F.
Diagnosis dapat ditegakkan
melalui beberapa pemeriksaan seperti hapusan darah tepi, sum-sum tulang dan
radiologis. Hitung jenis darah komplit menunjukkan adanya anemia dan rendahnya
MCV (mean corpuscular volume). Pemeriksaan elektroforesis bisa membantu, tetapi
tidak pasti, terutama untuk α-thalassemia. Karena itu diagnosis biasanya
berdasarkan kepada pola herediter dan pemeriksaan hemoglobin khusus.
TATALAKSANA
Pada thalassemia yang
berat diperlukan transfusi darah rutin dan pemberian tambahan asam folat. Pasien
yang menjalani transfusi, harus menghindari tambahan zat besi dan obat yang
bersifat oksidatif (misalnya sulfonamid), karena zat besi yang berlebihan bisa
menyebabkan keracunan. Pada bentuk yang sangat berat, mungkin diperlukan
pencangkokan sumsum tulang (HSCT/ Hematopoietic stem cell transplantation)
dengan mengikuti kriteria Lucarelli. Terapi
genetik masih dalam tahap penelitian.
Kelebihan besi merupakan
komplikasi fatal pada thalassemia bila tidak diatasi dengan baik, sehingga hal
ini menjadi fokus utama dalam tatalaksana thalassemia. Bila pasien tidak
mendapatkan kelasi besi (desferoxamine), akan terjadi disfungsi hati, jantung
dan kelenjar endokrin yang progresif
berakibat timbulnya fibrosis hati, sirosis hati, gagal jantung, diabetes
melitus, hipoganadism, hipotiroidism, hipoparatiroidism hinga kematian.
REFERENSI :
1.
Timan IS et al, Int J Hematol. 2002 Aug;76 Suppl 1:286-90
2.
Yuki, Thalassemia, USU, e-Repository, 2008
3.
Djajadiman G,dkk, Sari Pediatri, Vol.8, No.4, Mei 2007:78-84
4. What causes Thalassemias?, National Heart, Lung and Blood
Institute, https://www.nhlbi.ni.gov/health/health-topics/topics/thalassemia/causes.html
5. Whipple GH, Bradford WL. Mediteranean disease: thalassemia
(erythroblastic anemia of Cooley). Jpediatr. 1936;9:279-311.
Tidak ada komentar:
Posting Komentar